

来源于:原创 王昭月 生物探索
已有研究表明,某些分子能够增加癌细胞中肿瘤抑制蛋白PP2A的活性,从而杀死癌细胞以缩小细胞系和动物模型中的肿瘤。但其作用机制尚不清楚。基于此,密歇根大学的科学家们发现小分子DT-061可以特异性地稳定B56α-PP2A全酶,从而使其去磷酸化以达到抑癌效果。该研究成果发表在《cell》期刊上。
doi.org/10.1016/j.cell.2020.03.038
PP2A酶家族由三聚磷酸酶组成,即支架“A”亚基、催化“C”亚基和调节“B”亚基。其中,该蛋白的去磷酸化能力主要是由40多个决定特异性的调节“B”亚基竞争异质PP2A三聚体的组装和激活而调控的。通常一系列小分子PP2A激活剂(small molecule activators of PP2A,SMAPs)在一些体内肿瘤模型中可驱动c-Myc和ERK等特定致病底物的去磷酸化,从而协同抑制肿瘤。
小分子DT-061属于典型的SMAP,研究者利用细胞分离荧光素酶系统、免疫共沉淀(coIP)实验等,发现DT-061在细胞培养和体内都可以选择性地增强含有PP2A全酶种群的B56α。单独表达和纯化PP2A各亚基,发现DT-061可以直接稳定AB56αC异种三聚体。
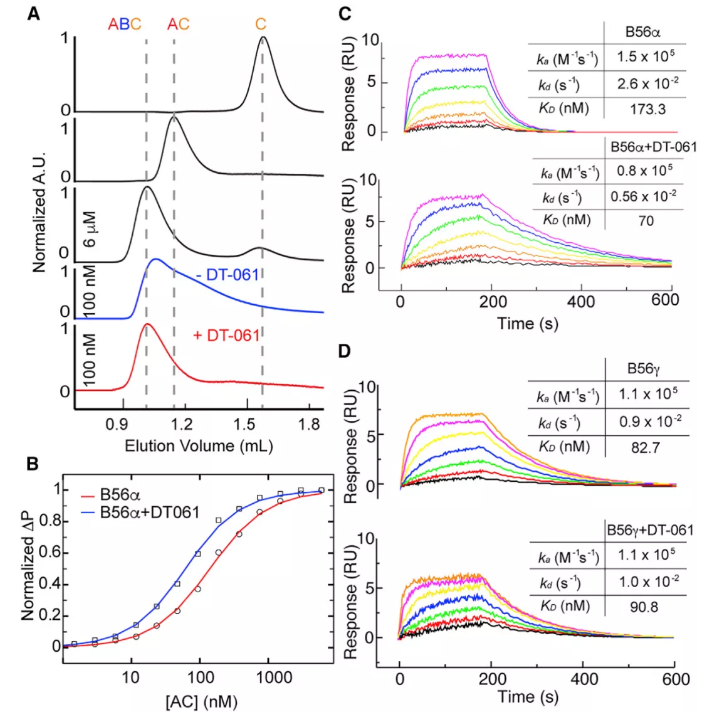
DT-061直接稳定AB56αC异种三聚体
然后利用单粒子低温电子显微镜(cryo-EM)来观察药物结合PP2A复合体的三维结构,发现DT-061结合在PP2A亚基间的独特“口袋”中,并与所有三个PP2A亚基的残基相互作用,其独特的稳定机制帮助我们揭示了一个难以捉摸和不对称的磷酸酶异质三聚体的结构。
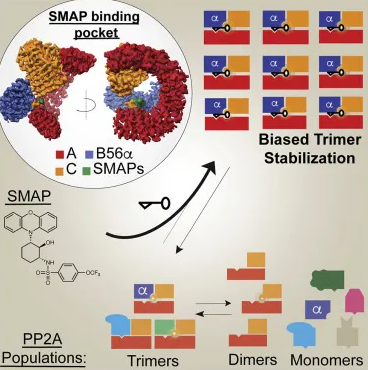
SMAPs选择性地稳定三亚单元以偏置磷酸酶PP2A
总之,研究者发现SMAPs结合在所有3个PP2A亚基间的“口袋”中,DT-061(SMAP)可以特异性稳定B56α-PP2A全酶,使后者可选择性地对底物如c-Myc去磷酸化。而甲基化的B56α异三聚体的积累可作为一种潜在的临床生物标志物。
研究者还表示,除了癌症,PP2A在心血管和神经退行性疾病等其他疾病中也出现失调现象,因此,该发现也为开发治疗心力衰竭和老年痴呆症等的新药提供了机会。
End
参考资料:
[1] Turning on the 'off switch' in cancercells.
[2] Selective PP2A Enhancement ThroughBiased Heterotrimer Stabilization, Cell (2020).